

Phenotype imputation increases effective sample size
We focused on the deepest available measure of MDD in UK Biobank11, LifetimeMDD, which we derived by applying clinical diagnostic criteria in silico to MDD symptom data from the Patient Health Questionnaire 9 (PHQ9) and the Composite International Diagnostic Interview Short Form (CIDI-SF) in the online Mental Health Questionnaire (MHQ). This procedure identified 16,297 LifetimeMDD cases and 50,867 controls. Because most individuals did not complete these questionnaires, LifetimeMDD was missing for 269,962 individuals. We also studied a shallow measure of MDD, GPpsy11, defined as seeking help from a general practitioner (GP) for “depression, anxiety, tension, or nerves”. For imputation and downstream analyses, we used a broad depression-relevant phenome with 217 phenotypes, including comorbidities and family history, as well as socioeconomic, demographic and environmental phenotypes (Supplementary Note and Supplementary Table 1).
We first imputed the depression phenome using SoftImpute24 (Methods), a variant of principal component analysis (PCA) that identifies latent factors from observed data and uses them to impute missing data. We previously found SoftImpute to be the most scalable among several established approaches17,24. We tuned SoftImpute’s regularization parameter using realistically held-out test data by taking unions of missingness patterns across samples17 and also used this approach to estimate the imputation accuracy for each phenotype (Extended Data Fig. 1)17. Imputation accuracy varied widely across phenotypes, ranging from R2 = 1% for being a twin to R2 = 97% for neuroticism score. For LifetimeMDD (80% missing), we estimated the phenotype imputation R2 to be 40%. This roughly translates to doubling its effective sample size17,25 (nobserved = 67,000, neffective = 166,000; Methods and Extended Data Fig. 1). Imputation accuracy was comparable when stratifying by sex, which is a significant MDD risk factor26,27,28 (Supplementary Fig. 1 and Supplementary Table 2). We found that the imputed measures had deflated variances and inflated correlations (Supplementary Note and Supplementary Fig. 2), as expected17. This effect could bias some downstream tests, such as tests for genetic correlation. One main goal of this work was to determine whether this approach to phenotype imputation is biased for large-scale single-trait genetic studies.
Finally, we applied a new deep-learning imputation method, AutoComplete29, to the same phenotype matrix (Methods). AutoComplete improved estimated imputation accuracy for most phenotypes with >10% missingness (29 of 42) and increased the average estimated by R2 by 2.9%.
Phenotype imputation improves GWAS power
We performed GWASs on observed LifetimeMDD (n = 67,164), imputed values of LifetimeMDD (ImpOnly, n = 269,962) and the concatenation of imputed and observed LifetimeMDD (ImpAll, n = 337,126; Fig. 2 and Methods). GWAS on the observed values of LifetimeMDD identified one significant locus (Fig. 2e). In GWASs on the imputed values, the number of GWAS loci increased to 13 and 18 for SoftImpute and AutoComplete, respectively (Fig. 2a, b and Supplementary Table 3). Finally, in GWASs on the combination of both imputed and observed values, the number of significant loci further increased to 26 and 40 for SoftImpute and AutoComplete, respectively (Fig. 2c, d and Supplementary Table 3). We confirmed that these improvements in the number of GWAS hits over the single hit from observed LifetimeMDD were unlikely to occur by chance (Supplementary Fig. 3).

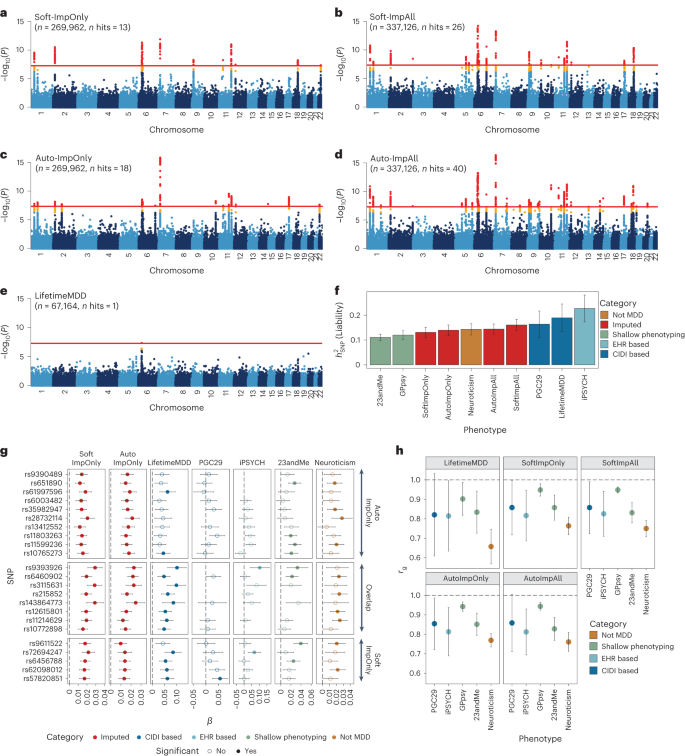
a–e, Manhattan plots for linear regression GWASs on imputed LifetimeMDD values from SoftImpute (a) and AutoComplete (c) (Soft-ImpOnly and Auto-ImpOnly, n = 269,962) and combined imputed and observed LifetimeMDD values from SoftImpute (b) and AutoComplete (d) (Soft-ImpAll and Auto-ImpAll, n = 337,126) and logistic regression GWAS on observed LifetimeMDD (e) (n = 67,164). −log10(P) values shown on the y axis were before adjustment for multiple testing; red lines show the genome-wide significance threshold of P < 5 × 10−8; P values and test statistics for all GWAS significant SNPs in a–e are shown in Supplementary Table 3. f, Liability-scale estimates of SNP-based heritability and h, genetic correlation between all UK Biobank phenotypes (n values for Soft, Auto and LifetimeMDD GWASs as above; GPpsy n = 332,629; neuroticism n = 274,056) and external MDD studies from PGC (n = 42,455), iPSYCH (n = 38,128) and 23andMe (n = 307,354). g, Replication of GWAS effect sizes from Soft-ImpOnly and Auto-ImpOnly in observed LifetimeMDD and external MDD studies. All error bars indicate 95% CI.
We investigated whether the new GWAS hits from phenotype imputation were MDD specific by comparing the ImpOnly GWASs to other MDD GWASs. First, we compared the two imputation methods. Of the 13 and 18 ImpOnly GWAS loci for SoftImpute and AutoComplete, respectively, 8 overlapped (giving a total of 23; Extended Data Fig. 2). Further, 9 of the remaining 15 loci had P < 10−5 in both ImpOnly GWASs, and all 15 of the 23 loci had P < 0.05/23. Overall, our two imputation methods captured highly overlapping genetic signals, but AutoComplete identified more loci. Next, we assessed the eight shared hits in four nonoverlapping depression cohorts (Methods and Supplementary Note): observed LifetimeMDD in UK Biobank, self-reported depression diagnosis or treatment in 23andMe7, the 29 MDD cohorts of the PGC4 (PGC29) and Danish registry data on MDD cases and population controls (iPSYCH15,30). For reference, we also compared our findings to those for neuroticism in UK Biobank, a personality trait that is genetically correlated with, but distinct from, MDD31. We found that all eight hits shared by both ImpOnly GWASs had sign-consistent effect sizes across all four depression cohorts, as well as in neuroticism. Moreover, all eight hits had P < 0.05/23 for observed LifetimeMDD in UK Biobank. Finally, of the 23 single-nucleotide polymorphisms (SNPs) significant in either of the ImpOnly GWASs, 18 replicated in at least one GWAS of observed MDD at P < 0.05/23 (Fig. 2g). Altogether, these results show that the loci underlying imputed LifetimeMDD are relevant to MDD.
We then checked whether the ImpOnly GWAS preserved the polygenic architecture of LifetimeMDD in terms of heritability and genetic correlation. First, we found that the liability-scale SNP-based heritability (\({h}_{{\rm{g}}({{\rm{liab}}})}^{2}\) from LDSC32) was lower for imputed LifetimeMDD (Soft-ImpOnly \({h}_{{\rm{g}}({{\rm{liab}}})}^{2}\) = 13.1%, standard error (SE) = 1.0%; Auto-ImpOnly \({h}_{{\rm{g}}({{\rm{liab}}})}^{2}\) = 14.0%, SE = 1.1%) than for observed LifetimeMDD (\({h}_{{\rm{g}}({{\rm{liab}}})}^{2}\) = 19.0%, SE = 2.9%; Fig. 2f). Nonetheless, the genetic correlations between imputed and observed LifetimeMDD were near 1 (Soft-ImpOnly: rg = 0.97, SE = 0.02; Auto-ImpOnly: rg = 0.96, SE = 0.03), as it was between the two imputation methods (rg = 1.00, SE = 0.004). Moreover, the rg between the ImpOnly phenotypes and other depression phenotypes largely mirrored the rg based on observed LifetimeMDD (Fig. 2h).
Finally, we tested for effect size heterogeneity between the ImpOnly and observed LifetimeMDD GWASs. We used a random-effect meta-analysis33 (Methods), as the ImpOnly and observed LifetimeMDD GWASs used nonoverlapping individuals. We found no significant heterogeneity in effect size between ImpOnly and observed LifetimeMDD at genome-wide significance (Extended Data Fig. 2), and across the 13 and 18 GWAS hits in Soft-ImpOnly and Auto-ImpOnly, respectively, 6 and 4 SNPs showed significant heterogeneity at P < 0.05/23. Altogether, our results show that imputed LifetimeMDD is noisier than observed LifetimeMDD but captures similar genetic effects.
Phenome-wide factors index pleiotropic axes of depression risk
We examined the top latent factors in SoftImpute to understand what phenotypic correlations drive the imputation. We used two statistical metrics to prioritize significant factors. First, we quantified the phenome-wide variance explained (Methods and Fig. 3a): the top handful of factors clearly stood out, with factors becoming comparable to background noise levels around factor 30. Second, we quantified factor stability by calculating the R2 between factors estimated on separate halves of the data, similar to prediction strength in clustering34 (Methods and Fig. 3b). We found that the first ten factors were very stable (min R2 of ~ 98%), with stability decaying steadily thereafter (factors 11–20, average R2 of ~ 80%; factors 21–30, average R2 of ~60%). We conservatively conclude that the first ten or so factors are statistically meaningful.

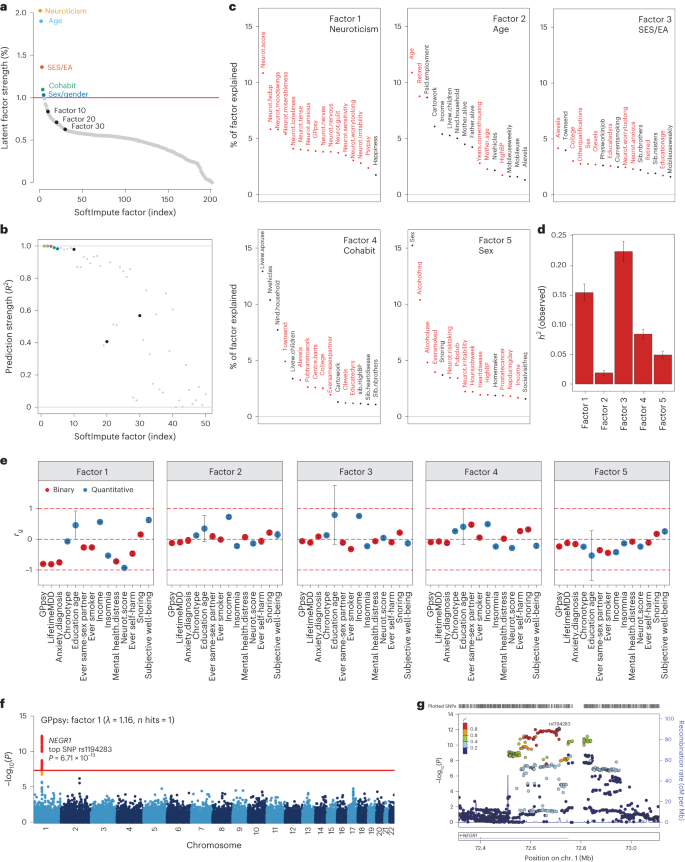
a,b, Statistical importance of each factor measured by percentage variance explained in the phenotype matrix (a) and factor prediction strength (b). c, Top phenotype loadings for the top five SoftImpute factors. d,e, Estimates of heritability (d) (n = 337,126 for all factors) and genetic correlation (e) of the top five SoftImpute factors with MDD-relevant traits (n values for all MDD-relevant traits can be found in Supplementary Table 1). f, Logistic regression GWAS Manhattan plot of GPpsy conditioning on SoftImpute factor 1; the red line shows the genome-wide significance threshold. g, LocusZoom plot of the significant GWAS locus in gene NEGR1. All error bars indicate 95% CI.
We studied the genetic basis of each factor with GWASs. The number of GWAS hits ranged from 3 (factor 2, mainly indexing age-related phenotypes; Fig. 3c) to 309 (factor 3, mainly indexing socioeconomic status and education attainment), with λGC ranging from 1.15 (factor 2) to 2.11 (factor 3) (Supplementary Fig. 5). We next estimated heritabilities and found they ranged from \({h}_{{\rm{g}}}^{2}\) = 1.9% (SE = 0.2%) for factor 2 to \({h}_{{\rm{g}}}^{2}\) = 22.4% (SE = 0.9%) for factor 3 (Fig. 3d and Supplementary Fig. 4). Finally, we profiled the genetic correlation between factors and MDD-related phenotypes (Fig. 3e and Supplementary Fig. 4). We found that the genetic correlations closely mirrored the factor loadings, which are based on phenotypic correlations. For example, factor 1 had rg = −0.93 (SE = 0.01) with neuroticism and factor 3 was correlated with years of education (rg = 0.79 (SE = 0.96) and income (rg = 0.75, SE = 0.03).
Given these results, we hypothesized that our top phenome-wide factors partly capture nonspecific pathways contributing to shallow MDD phenotypes. To test this hypothesis, we performed GWAS analysis on a shallow MDD measure, GPpsy (n = 332,629), conditional on factor 1. This is akin to removing confounders such as batch effects in eQTL studies through conditioning on latent factors. We found that only 1 of the 25 GWAS hits for GPpsy remained after adjusting for factor 1 (Fig. 3f). This hit overlaps NEGR1 (top SNP rs1194283, odds ratio (OR) = 1.05, SE = 0.0065, P = 6.71 × 10−13; Fig. 3g), which has been identified as an MDD risk locus in multiple GWASs with varying phenotyping approaches4,6,8,35,36,37. Intriguingly, this locus also has replicated associations with body mass index and obesity in multiple populations38,39,40,41. We also performed GWASs conditioned on each of the other top ten factors, which generally had little impact (Supplementary Fig. 6). One clear exception, however, was that adjusting for factor 3 increased the number of GPpsy GWAS hits from 25 to 35. These additional loci could be false positives from collider bias23,42,43.
MTAG improves GWAS power but is sensitive to inputs
As an alternative to phenotype imputation, we next evaluated phenotype integration at the GWAS summary statistic level using MTAG19, an inverse-covariance-weighted meta-analysis for GWAS on multiple traits. We did not use all 217 phenotypes in MTAG for two reasons. First, MTAG requires GWAS to be run on each input phenotype, which is computationally intractable for hundreds of phenotypes. Second, MTAG produces false positives when applied to large numbers of input GWAS19. Instead, we evaluated six different sets of input GWAS to MTAG, producing six different integrated LifetimeMDD GWASs (Fig. 4a, Extended Data Fig. 3, Supplementary Table 4 and Supplementary Note).

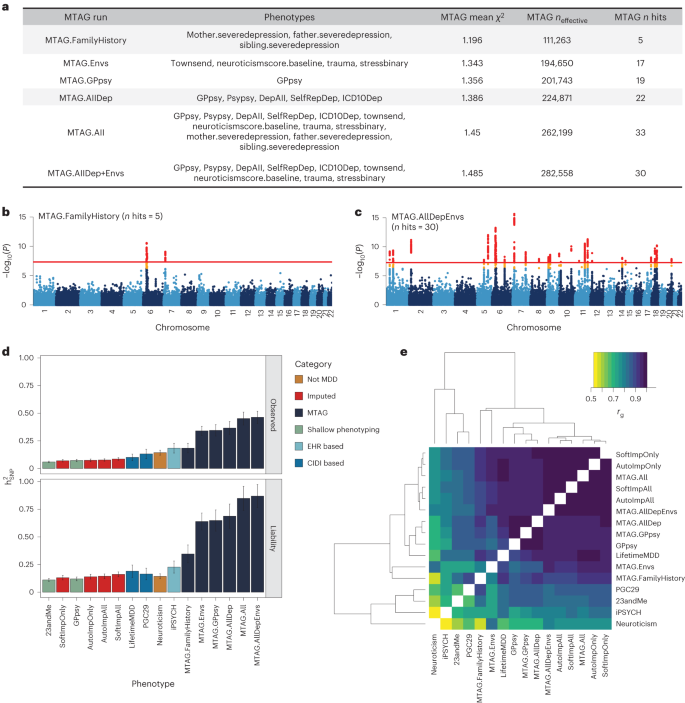
a, Description of the evaluated input choices for MTAG and their resulting GWAS summary statistics. The MTAG effective sample size refers to the power-equivalent sample size of the MTAG GWAS versus the single-trait GWAS19, calculated as neffect = nsingle ⨯ (χ2MTAG − 1)/(χ2single − 1), where the χ2 terms are the average GWAS chi-squared values. b,c, Manhattan plots for the MTAG models with the fewest (MTAG.FamilyHistory) (b) and most (MTAG.AllDep+Envs) (c) GWAS hits. −log10(P) values shown on the y axis were before adjustment for multiple testing; red lines show the genome-wide significance threshold of P < 5 × 10−8. d, SNP-based heritability estimates on the observed and liability scales for observed, imputed, and MTAG GWASs on LifetimeMDD as well as reference phenotypes (neffect is shown in Supplementary Table 5). e, Estimated genetic correlations for observed, imputed and MTAG analyses of LifetimeMDD and reference phenotypes. All error bars indicate 95% CI.
All six MTAG input choices increased the number of GWAS hits from observed LifetimeMDD. On the low end, MTAG using family history measures of depression yielded five GWAS hits (MTAG.FamilyHistory, λGC = 1.20; Fig. 4b). On the high end, MTAG using shallow MDD phenotypes and environmental factors (such as recent stressful life events, lifetime traumatic experiences and the Townsend deprivation index) yielded 33 GWAS hits (MTAG.All, λGC = 1.45; Fig. 4c). We note that MTAG.AllDep is analogous to prior depression phenotypes defined by manually combining similar depression measures14 and that MTAG.FamilyHistory is analogous to prior methods that integrated family history into GWAS analysis44,45. Of the 51 total hits across all MTAG runs, 34 overlapped hits from the imputed GWASs (Extended Data Fig. 3). Notably, we found that adding more input phenotypes in MTAG always increased the number of GWAS hits. This reflects a combination of added power by leveraging pleiotropy and an increased false positive rate19. Additionally, MTAG GWASs yielded substantially inflated heritability estimates (on both the liability and observed scales), which increased with more input GWAS. For example, MTAG.All gave \({h}_{{\rm{g}}({{\rm{liab}}})}^{2}\) = 84.9% (SE = 5.6%), compared to \({h}_{g({liab})}^{2}\) = 19.0% (SE = 2.9%) for observed LifetimeMDD (Fig. 4d and Supplementary Table 5).
We next examined genetic correlations between MTAG and other MDD GWASs (Fig. 4e). First, MTAG.All, which included the most input GWAS, clustered together with the imputed GWASs, which leveraged all 217 phenotypes. Second, MTAG using shallow MDD phenotypes (MTAG.AllDep and MTAG.GPpsy) clustered with the GWAS on GPpsy. Third, neuroticism was significantly more genetically correlated with MTAG.Envs (rg = 0.84, SE = 0.01) than LifetimeMDD (\({r}_{g}\) = 0.66, SE = 0.06). These results are consistent with prior observations that MTAG-based summary statistics modestly inflate genetic correlation with the input phenotypes46. Overall, the genetic correlations between MTAG and LifetimeMDD were high, with the lowest value derived for MTAG.Envs (rg = 0.90, SE = 0.03).
Finally, to compare like to like, we evaluated SoftImpute’s accuracy using only the MTAG.All input phenotypes (plus sex, age and 20 PCs). We found that imputation performed much worse with this reduced set of phenotypes (phenome-wide R2 decreased from 59.6% to 39.5%, P < 2 × 10−5, pooled t-test across folds; Supplementary Fig. 2 and Supplementary Table 2).
Phenotype imputation and MTAG improves PRS accuracy
We then assessed the within-sample prediction accuracy of PRSs built from integrated MDD GWAS. We used ten-fold cross-validation to obtain the Nagelkerke’s \({R}^{2}\) prediction accuracy for LifetimeMDD in white British individuals in UK Biobank. We jointly cross-validated the phenotype imputation and PRS construction (Methods). For MTAG, we jointly cross-validated the GWASs on secondary input phenotypes. For context, we compared these PRSs to ones built from observed LifetimeMDD (n = 67,164; for neffect see Supplementary Table 6) and GPpsy (n = 332,629) in UK Biobank11, MDD defined by structured interviews in PGC29 (ref. 4) (n = 42,455), affective disorder defined by Danish health registries in iPSYCH15 (n = 38,123) and self-reported depression in 23andMe7 (n = 307,354; Supplementary Table 6).
We found that imputing LifetimeMDD doubled PRS prediction accuracy over observed LifetimeMDD (Fig. 5a; LifetimeMDD: R2 = 1.0%, 95% confidence interval (CI) = 0.6–1.4%; Soft-ImpAll: R2 = 2.1%, 95% CI = 1.3–2.9%; Auto-ImpAll: R2 = 2.2%, 95% CI = 1.4–3.0%). Consistent with prior reports11,12, we found that the GPpsy PRS predicted LifetimeMDD better than the LifetimeMDD PRS itself (R2 = 1.6%, 95% CI = 0.6%–2.4%) because it has a roughly four times larger sample size. Nonetheless, both the SoftImpute and AutoComplete PRSs outperformed the GPpsy PRS, demonstrating that integrating shallow and deep phenotypes through imputation can improve PRS accuracy over that with either class of phenotypes alone. Finally, we found that the imputed LifetimeMDD PRS substantially outperformed the PRSs from iPSYCH (R2 = 0.6%, 95% CI = 0.2–0.9%) and 23andMe (R2 = 1.3%, 95% CI = 0.7–1.9%), even though iPSYCH used deeper phenotypes and 23andMe had a large sample size.

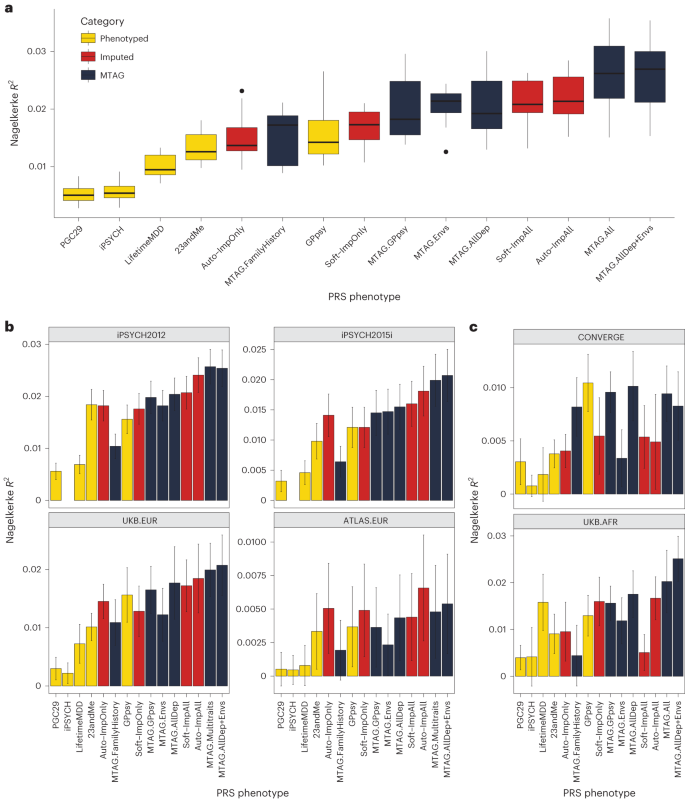
a, PRS prediction accuracy for LifetimeMDD (n = 67,164) in unrelated white British individuals in UK Biobank using ten-fold cross-validation. For the imputed PRSs, we also cross-validated the imputation. Median values are shown as a line in the box; whiskers of boxplots are 1.5 times the interquartile range; outliers outside of the interquartile range are shown as filled dots. b, Out-of-sample PRS prediction accuracy in four additional cohorts with European ancestries (iPSYCH2012, n = 42,250; iPSYCH2015i, n = 23,351; UKB.EUR, n = 10,193; ATLAS.EUR, n = 14,366). c, PRS prediction accuracy in African-ancestry individuals in UK Biobank (n = 687) and Han Chinese-ancestry individuals in CONVERGE (n = 10,502). All error bars indicate 95% CI.
The performance of the MTAG PRSs mirrored the MTAG GWAS results and depended on the input phenotypes (Fig. 4 and Extended Data Fig. 3). First, MTAG.FamilyHistory showed improved accuracy compared to observed LifetimeMDD but underperformed in comparison to the imputed PRSs (R2 = 1.5%, 95% CI = 0.6–2.5%; Fig. 5a). On the other hand, MTAG.All outperformed the imputed PRSs by about 20% (R2 = 2.6%, 95% CI = [1.3%, 3.9%]; Fig. 5a). In particular, this demonstrates that MTAG with more than ten inputs, which is nonstandard and likely yields miscalibrated GWAS results, can significantly improve PRS prediction.
Phenotype imputation and MTAG improves PRS portability
We next asked whether phenotype integration improves PRS predictions in different cohorts, diagnostic systems and/or populations. Demonstrating portability is essential to establish that phenotype integration does not merely reflect dataset-specific biases.
First, we tested PRS accuracy in non-British European-ancestry individuals in UK Biobank (UKB.EUR, n = 10,166). These individuals were measured on the same LifetimeMDD phenotype as our sample of white British UK Biobank individuals and also have European ancestry, and hence represent the most similar cohort (Supplementary Note and Supplementary Fig. 7). Although the small sample size limits definitive conclusions, we observed a nearly identical pattern among the PRS methods as in our training sample: imputation and MTAG almost always improved prediction accuracy compared to both LifetimeMDD and GPpsy (Fig. 5b). We next assessed portability to two large European-ancestry cohorts from iPSYCH (2012 cohort (n = 42,250) and 2015i cohort (n = 23,351); Supplementary Note). These nonoverlapping samples were drawn from a nationwide Danish birth cohort with diagnoses obtained from national health registers30,47. We again found qualitatively identical results, with imputation outperforming both LifetimeMDD and GPpsy and the best MTAG setting outperforming imputation (Fig. 5b). Finally, we tested portability to European-ancestry individuals in the ATLAS dataset based on MDD as defined in University of California – Los Angeles electronic health record (EHR) data48,49 (ATLAS.EUR, n = 14,388; Supplementary Note, Supplementary Fig. 8 and Supplementary Tables 6–8). Again, the small sample size prevented definitive comparisons, but phenotype imputation and the best MTAG setting improved estimated accuracy (Fig. 5b).
We next tested these PRSs in individuals with non-European genetic ancestries, including African-ancestry individuals7 in UK Biobank with observed LifetimeMDD status (UKB.AFR, n = 687), as well as Han Chinese-ancestry individuals in the CONVERGE cohort9,50 (n = 10,502; Supplementary Note) who were assessed for severe, recurrent MDD (Fig. 5c, Supplementary Note and Supplementary Table 6). Consistent with previous studies51,52,53, we found that the PRSs we derived from GWASs on European-ancestry cohorts generally had poorer portability to non-European-ancestry cohorts, making firm conclusions difficult. Nonetheless, the best MTAG setting was always nearly optimal, and PRSs based on imputed LifetimeMDD always outperformed the PRS based on observed LifetimeMDD. Finally, we tested PRS prediction accuracy in UK Biobank individuals with Asian ancestry (UKB.ASN, n = 334), as well as ATLAS individuals who self-identified as Latino (ATLAS.LAT, n = 2,454), Black (ATLAS.AFR, n = 1,158) or Asian (ATLAS.ASN, n = 1,996). However, the power was too low in these small cohorts for meaningful interpretation (Supplementary Fig. 9).
A new metric for the specificity of PRSs
While phenotype integration improves PRS prediction in UK Biobank and in external cohorts, this may come at the cost of reduced specificity to MDD. This is because integration explicitly borrows information from secondary phenotypes, which could introduce genetic signals that are not specific to MDD. To quantify this spillover of nonspecific effects into an MDD PRS, we compared prediction accuracy for LifetimeMDD to prediction accuracy for secondary phenotypes. We call this metric of specificity ‘PRS pleiotropy’ (R2secondary/R2LifetimeMDD). Because core MDD biology is likely partly pleiotropic, its PRS pleiotropy should be nonzero for many secondary phenotypes. We further expect that shallow MDD phenotypes, such as GPpsy, would generally have higher PRS pleiotropy than LifetimeMDD11.
For all PRSs based on observed and integrated MDD, we calculated PRS pleiotropy for 172 secondary phenotypes used in imputation. We then investigated the 62 secondary phenotypes that were significantly predicted by any examined PRS (P < 0.05/172; Methods). Visualizing PRS pleiotropy for observed LifetimeMDD across this depression phenome showed a spectrum of highly linked traits, including shallow MDD phenotypes such as GPpsy and genetically correlated traits such as neuroticism, that quickly faded across successive phenotypes (Fig. 6a). By comparison, GPpsy broadly had higher PRS pleiotropy across secondary phenotypes, indicating that GPpsy captures less-specific biology than LifetimeMDD, as expected. We also found that the 23andMe GWAS had similar PRS pleiotropy to GPpsy, consistent with the fact that both measure MDD by self-reported depression.

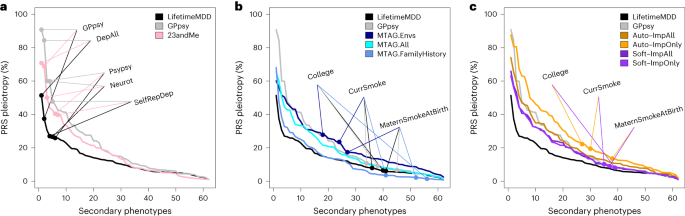
PRS pleiotropy spectra across the depression-relevant phenome, defined as the ratio of PRS prediction accuracy for secondary traits relative to LifetimeMDD (PRS pleiotropy = R2secondary/R2LifetimeMDD). a, PRSs derived from GWAS on shallow MDD phenotypes (GPpsy or 23andMe) are less specific to LifetimeMDD than the PRS derived from the GWASs on LifetimeMDD. b, MTAG-based PRSs range from highly specific (MTAG.FamilyHistory) to less specific (MTAG.Envs) compared to PRSs derived from shallow MDD phenotypes. c, SoftImpute PRSs are more specific than PRSs derived from shallow MDD phenotypes, while AutoComplete PRSs are similar. Note that GPpsy and LifetimeMDD are each used in two ways: (1) to build the PRS and (2) to evaluate PRS pleiotropy.
Our main question here was whether phenotype integration has more PRS pleiotropy than self-reported depression as in GPpsy and 23andMe. We first evaluated PRS pleiotropy for MTAG and found that specificity highly depended on the input GWAS (Fig. 6b). First, MTAG.Envs had far higher PRS pleiotropy than GPpsy, showing that its high PRS power comes at a high cost to specificity. On the other hand, MTAG.All had a similar PRS pleiotropy as GPpsy and nearly double the PRS R2; hence, MTAG.All is clearly superior to GPpsy. MTAG.FamilyHistory had the opposite properties: it only modestly improved PRS R2 over observed LifetimeMDD, but this benefit came without loss of specificity. We then evaluated PRS pleiotropy for imputed phenotypes (Fig. 6c). The SoftImpute ImpAll and ImpOnly PRSs were both more specific to LifetimeMDD than the GPpsy PRS, which is notable given that the imputed values were constructed from more than 200 phenotypes, including GPpsy. The AutoComplete ImpOnly PRS was less specific than GPpsy, although the ImpAll PRS was comparable.
Finally, we evaluated PRS pleiotropy relative to observed LifetimeMDD to ask which of the phenotypes had excess pleiotropy in the integrated PRSs. We defined ‘excess PRS pleiotropy’ as the PRS pleiotropy minus the PRS pleiotropy of observed LifetimeMDD11. As expected, self-reported depression PRSs (GPpsy and 23andMe) had high excess PRS pleiotropy, especially when compared to shallow MDD measures (Extended Data Fig. 4a). Likewise, MTAG.Envs had substantial excess PRS pleiotropy, especially for socioeconomic measures such as years of education (Extended Data Fig. 4b). Notably, MTAG.FamilyHistory had far less excess PRS pleiotropy than other MTAG settings or GPpsy, as well as reduced pleiotropy for several socioeconomic measures. Finally, SoftImp-All had lower excess PRS pleiotropy than GPpsy (41/62 phenotypes); however, AutoImp-All had higher excess PRS pleiotropy (Extended Data Fig. 4c). Overall, MTAG can outperform imputation in PRS sensitivity or specificity depending on input, while imputation provides a simple and scalable approach that performs well in both power and specificity.
We then downsampled each GWAS used to build a PRS to assess the impact of sample size (Supplementary Note). Overall, we found that PRS pleiotropy was stable, although it can be upwardly biased for sample sizes below 100,000 (due to low power). In particular, our results were robust to differences in the training PRS sample sizes, except that observed LifetimeMDD PRS pleiotropy is a conservative baseline because it is trained on 67,000 individuals (Extended Data Fig. 5 and Supplementary Figs. 10 and 11). Further, we confirmed that these results persisted when we used the same SNPs in each PRS (Supplementary Note and Extended Data Fig. 6).







